Select Publications
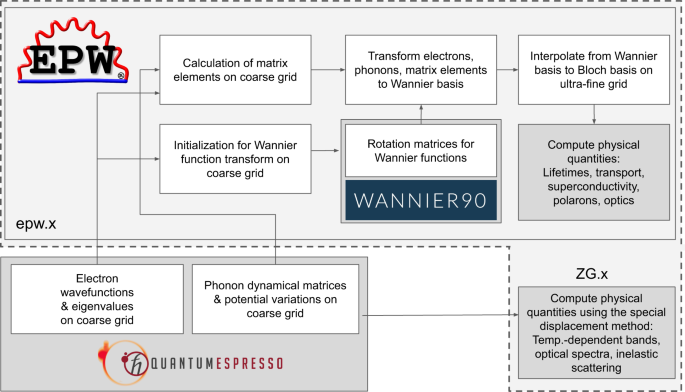
Electron-phonon physics from first principles using the EPW code
H. Lee, S. Poncé, K. Bushick, S. Hajinazar, J. Lafuente-Bartolomé,
J. Leveillee, C. Lian, J.-M. Lihm, F. Macheda, H. Mori, H. Paudyal, W. H. Sio,
S. Tiwari, M. Zacharias, X. Zhang, N. Bonini, E. Kioupakis, E. R. Margine, and F. Giustino,
npj Comput. Mater. 9, 156 (2023)
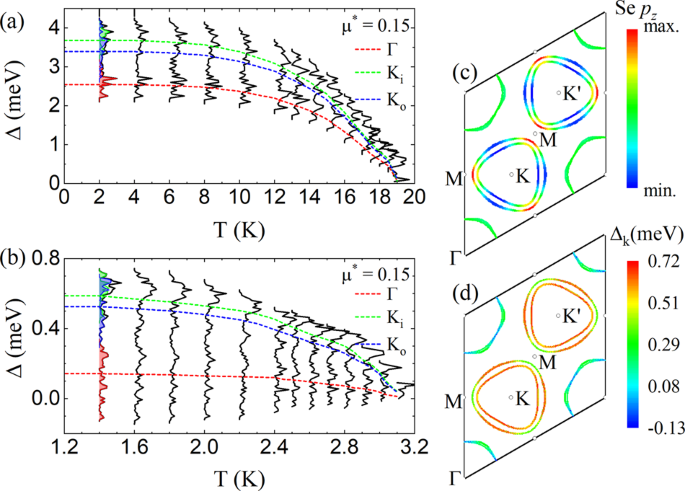
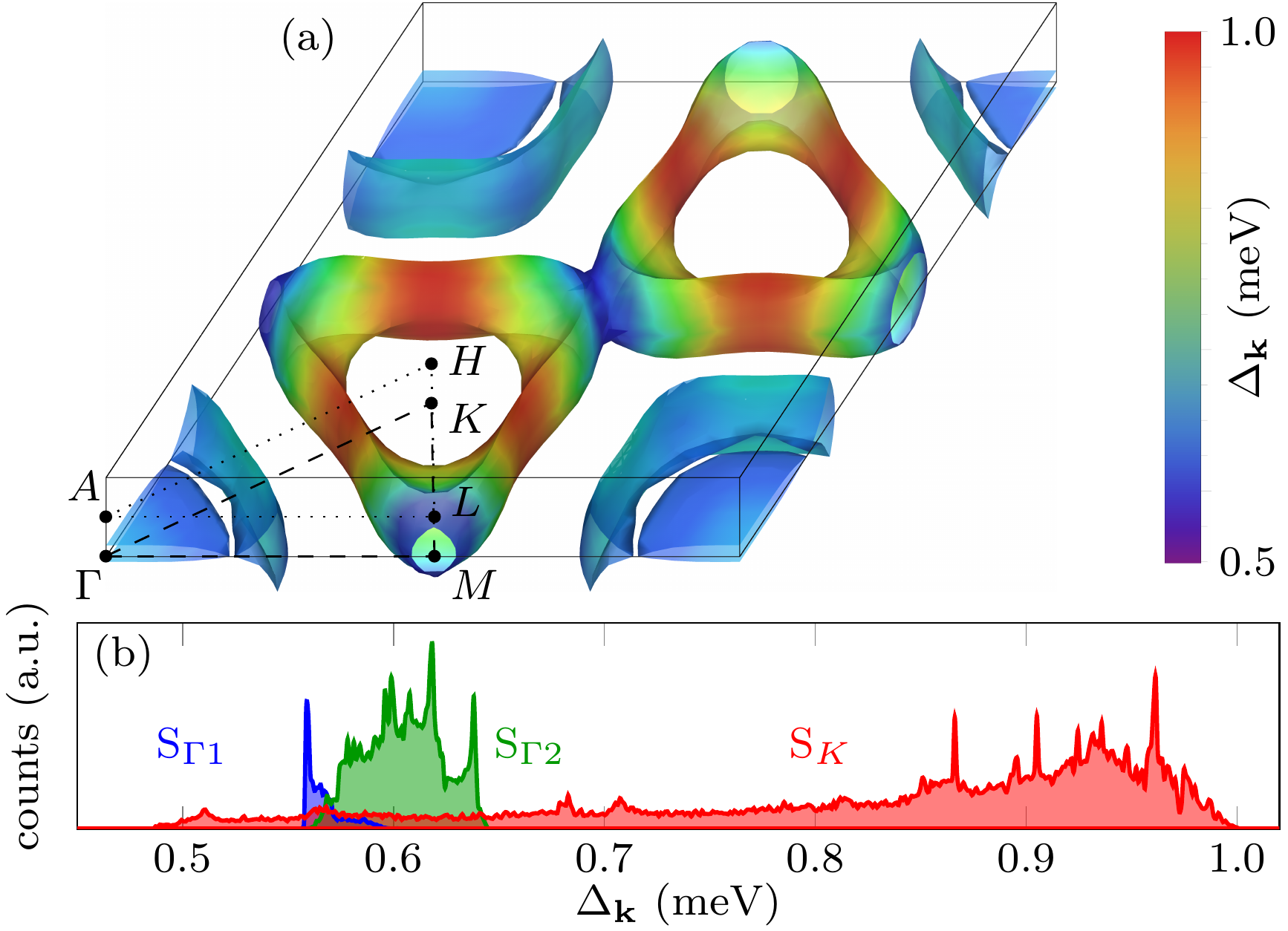
Electron-phonon coupling and spin fluctuations in the Ising superconductor NbSe2
S. Das, H. Paudyal, E. R. Margine, D. F. Agterberg, and I. I. Mazin,
npj Comput. Mater. 9, 66 (2023)
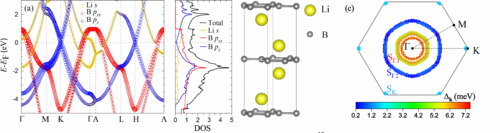
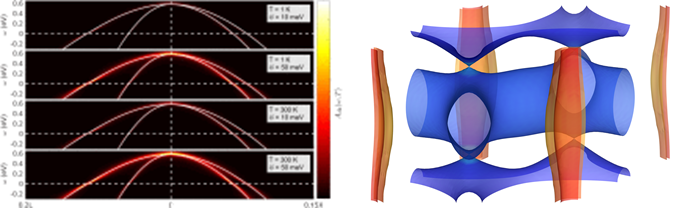
Ab initio study of Li-Mg-B superconductors
G. P. Kafle, C. R. Tomassetti, I. I. Mazin, A. N. Kolmogorov, and E. R. Margine,
Phys. Rev. Materials 6, 084801 (2022)
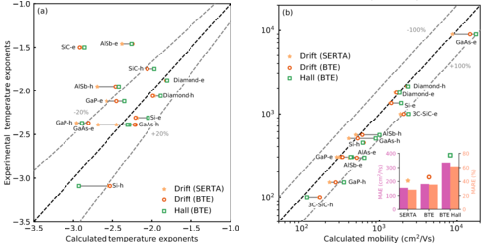
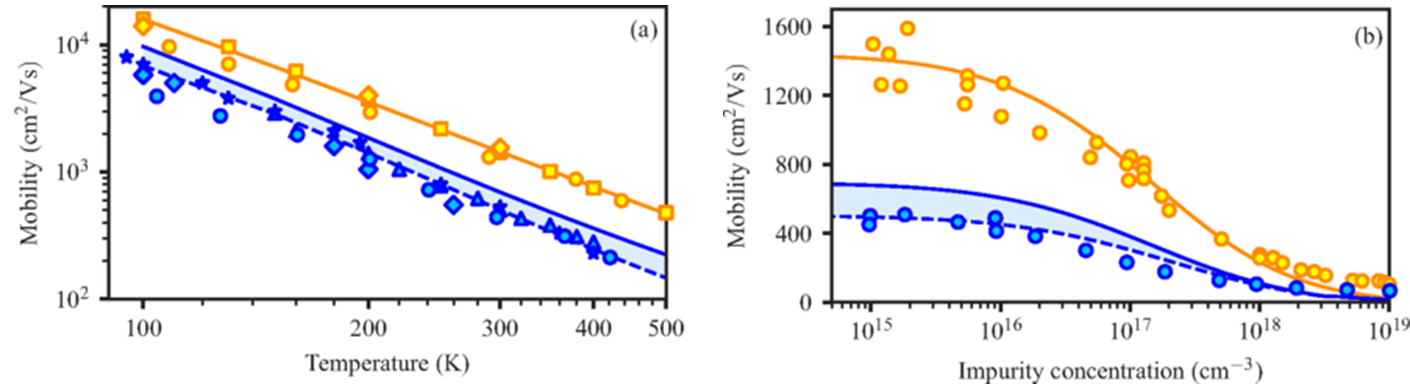
First-principles predictions of Hall and drift mobilities in semiconductors
S. Poncé, F. Macheda, E. R. Margine, N. Marzari, N. Bonini, and F. Giustino,
Phys. Rev. Research 3, 043022 (2021)
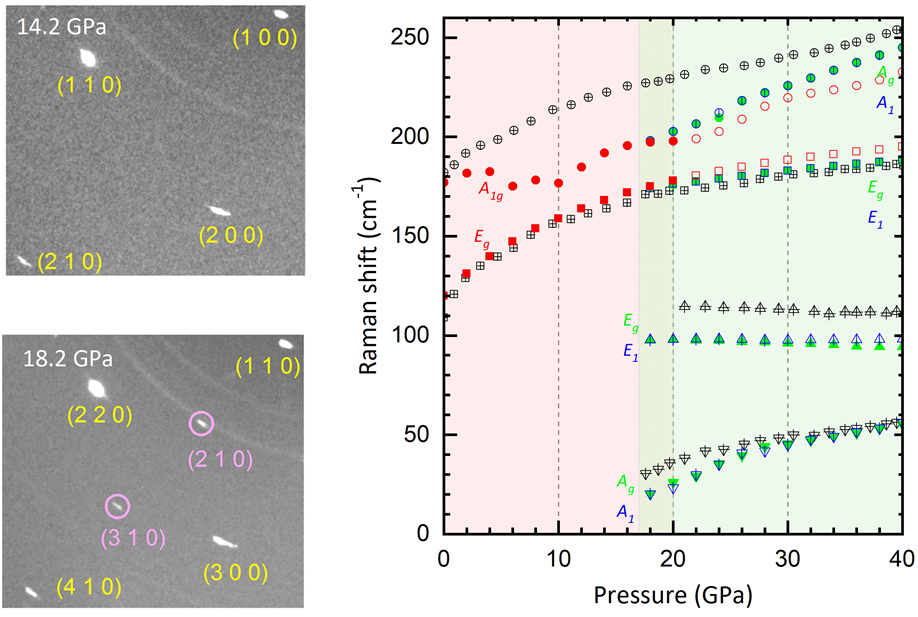
Unusual Pressure-Induced Periodic Lattice Distortion in SnSe2
J. Ying, H. Paudyal, C. Heil, X.-J. Chen, V. V. Struzhkin, and E. R. Margine,
Phys. Rev. Lett. 121, 027003 (2018)
Towards predictive many-body calculations of phonon-limited carrier mobilities in semiconductors
S. Poncé, E. R. Margine, and F. Giustino, Phys. Rev. B 97, 121201(R) (2018)
Origin of Superconductivity and Latent Charge Density Wave in NbS2
C. Heil, S. Poncé, H. Lambert, M. Schlipf, E. R. Margine, and F. Giustino,
Phys. Rev. Lett. 119, 087003 (2017)
EPW: Electron–phonon coupling, transport and superconducting properties using maximally localized Wannier functions
S. Poncé, E. R. Margine, C. Verdi, and F. Giustino, Comput. Phys. Commun. 209, 116 (2016)
Research Sponsors